- Paper link ペイパーリンク
- 読むにあたっての最低限の知識 The minimum requirement to read this paper.
- 分散共分散行列と多変量正規分布 Variance-covariance matrix and multi-variate normal distribution
- 分散共分散行列は、個体間の「近さ」を成分とした行列。標準化すると、「一致」することは1となる
- Variance-covariance matrix is a matrix with elements representing similarity among samples. Standardized matrix has 1 for identitiy.
- 対角成分は「自身」と「自身」との関係だから「一致」
- Diagonal elements stand for self-self relation (identical each other).
- 多変量正規分布は、分散共分散行列によって、個体間の近さが指定される
- Multi-variate normal distribution has a variance-covarance matrix that specifies similarities among samples.
- 以下のRコマンドによって、以下のことが確かめられる You can learn the following items with R commands below.
- 血縁関係が遺伝因子による分散共分散行列成分を定めること Kinship determines values of genetic variance-covariance matrix.
- 遺伝率は、遺伝因子による分散共分散行列成分と、非遺伝因子による分散共分散行列成分との比率であること Heritability is the fraction of genetic matrix and non-genetic matrix.
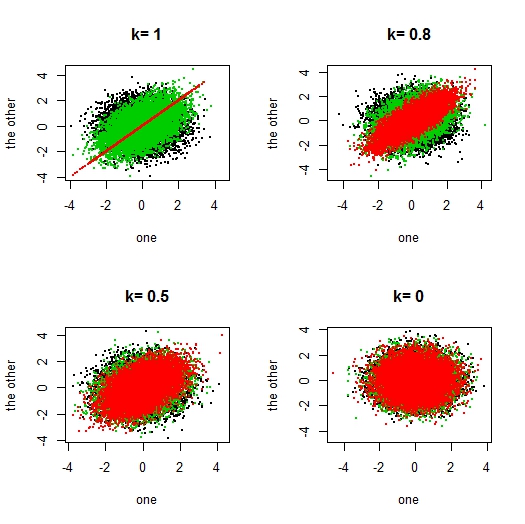
- そのようにして作成した分散共分散行列に基づいて生成される多変量正規分布乱数では、確かに、血縁関係が強い個人同士の表現型値は近くなること The total variance-covariance matrix (sum of two matrices) gives random vectors from multi-variate normal distribution and the values of samples in close kinships tend to have close phenotype values.
# install.packages("mvtnorm")
library(mvtnorm)
# 遺伝的な関係の強さを表す行列を作る Make a matrix G based on kinship
n <- 6 # 個体数 no. samples
G <- diag(rep(1,n)) # 自身との相関は1 Self-self identity
G
# 血縁関係に応じて相関の値を与える Specify values based on kinship
G[1,2] <- G[2,1] <- 1 # s1,s2は一卵性双生児 monozygotic twin
G[3,4] <- G[4,3] <- 0.5 # s3,s4は二卵性双生児 dizygotic twin
G[5,6] <- G[6,5] <- 0.02 # s5,s6は少し血縁関係がある a bit related
# (s1,s2),(s3,s4),(s5,s6)以外の関係は全くの無関係 no relation between pairs except for (s1,s2),(s3,s4),(s5,s6)
G
# こんな6個体をn.iter回、集めてみることにする Observe these 6 individuals for n.iter times.
n.iter <- 10^4
# 多変量正規分布に従う乱数を発生させる Random generation of 6 sample phenotype values
# 遺伝要因以外の寄与分は、独立(非対角成分は0)とする Non-genetic matrix is for independence model for here.
E <- diag(rep(1,n))
# 遺伝率 k heritability
# 遺伝率は、GとEとの割合のこと heritability is fraction of G out of G+E
par(mfrow=c(2,2))
ks <- c(1,0.8,0.5,0) # 4通りの遺伝率を試す Four k values
Vs <- Xs <- list()
for(i in 1:length(ks)){
Vs[[i]] <- ks[i]*G + (1-ks[i])*E
Xs[[i]] <- rmvnorm(n.sample,sigma=Vs[[i]])
plot(Xs[[i]][,5],Xs[[i]][,6],xlim=range(Xs[[i]]),ylim=range(Xs[[i]]),cex=0.1,pch=20,xlab="one",ylab="the other",main=paste("k=",ks[i]))
points(Xs[[i]][,3],Xs[[i]][,4],cex=0.1,pch=20,col=3)
points(Xs[[i]][,1],Xs[[i]][,2],cex=0.1,pch=20,col=2)
}
par(mfcol=c(1,1))
- 論文の概要 Paper's Abstract
- 遺伝率の推定は、個人の表現型のばらつきを遺伝因子による部分とそれ以外の部分との和に分け、遺伝因子による部分の割合を推定すること
- Estimation of heritability is estimation of fraction of genetic variance after dividing phenotype variance into the sum of genetic and non-genetic variances.
- 端的に表せば Its formula:
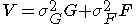
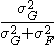
- のように分けること。ただしGとFとはそれぞれ個人同士の遺伝的相関、非遺伝的相関を表す行列で、各列が平均0、分散1に標準化してあるもの
- Divide into two terms as above; G and F are correlation matrices among samples for genetic and the rest whose mean and variance are normalized.
- 双生児データや家系データの場合にはGの成分として、血縁関係に応じた値(一卵性双生児なら1、二卵性双生児なら0.5など)を使い、それに基づいて、観測表現型から遺伝率を推定する
- In case of twin and familial data studies, the values of matrix G can be given based on samples' kinship, such as 1 for monozygotic twins and 0.5 for dizygotic twins, and heritability is estimated from the observed phenotype values.
- ゲノムワイドSNPデータがあれば、血縁関係がないとみなせる諸個人間の遺伝的な近さが数値にできるので、それを用いてGを作って遺伝率を推定しよう、というのが本論文の内容
- With genomewide SNP data, genetic correlation among non-familial, populational samples can be estimated, which enables us to make G matrix, subsequently we can estimate heritability.

コメントをかく