LAMMPS version: lammps-17Nov16
CUDA version : CUDA-8.0 (newest at 2016/11/23)
mpirun -np 8 /home/qolo/calc_tools/lammps-17Nov16/src/lmp_mpi -sf gpu -pk gpu 1 neigh no -i input.in > output.log &
you cannot use pppm(Particle-Particle-Particle-Mesh ewald) in triclinic geometry box. if the box, use "kspace_style ewald X.XXXX", alternatively.
package option have to be included in command.
check command for work of GPU: nvidia-smi
Examples:
INSTALLATION memo ( lib/gpu/Makefile.your-choice );
NVCC = /usr/local/cuda-8.0/bin/nvcc -Xcompiler -fPIC
CUDA_ARCH = -arch=sm_61 I use GTX 1060, check "Compute ability" at https://en.wikipedia.org/wiki/CUDA
CUDA version : CUDA-8.0 (newest at 2016/11/23)
mpirun -np 8 /home/qolo/calc_tools/lammps-17Nov16/src/lmp_mpi -sf gpu -pk gpu 1 neigh no -i input.in > output.log &
you cannot use pppm(Particle-Particle-Particle-Mesh ewald) in triclinic geometry box. if the box, use "kspace_style ewald X.XXXX", alternatively. 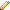
package option have to be included in command.
check command for work of GPU: nvidia-smi
Examples:
- -----------------------------------------------------------------------------+
| Processes: GPU Memory |
| GPU PID Type Process name Usage |
| ============================================================================= |
| 0 1062 G /usr/lib/xorg/Xorg 153MiB |
| 0 1887 G /usr/bin/compiz 42MiB |
| 0 4852 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 73MiB |
| 0 4853 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 71MiB |
| 0 4854 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 73MiB |
| 0 4855 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 73MiB |
| 0 4856 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 71MiB |
| 0 4857 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 71MiB |
| 0 4858 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 73MiB |
| 0 4859 C ...olo/calc_tools/lammps-17Nov16/src/lmp_mpi 73MiB |
- -----------------------------------------------------------------------------+
INSTALLATION memo ( lib/gpu/Makefile.your-choice );
NVCC = /usr/local/cuda-8.0/bin/nvcc -Xcompiler -fPIC
CUDA_ARCH = -arch=sm_61
I use GTX 1060, check "Compute ability" at https://en.wikipedia.org/wiki/CUDA
LMP_INC = -DLAMMPS_SMALLBIG -D_FORCE_INLINES
''Cuda driver error 4 in call at file 'geryon/nvd_device.h' in line 124.''
This Error message distressed me ... :(
following text is very old version. you should see the newest official manual
本テキストはCentOS 5.10で行っています。gccなどの基本的なパッケージは最初から入っているため、以下では省いています。Windowsはバイナリが用意されていた(ような気がする)なので、OpenMPを使う場合はすぐに計算をすることができます(遅いけど)。Cygwin環境と併せていずれ機会があれば書いてみたいなぁとは思っていますが、今回はLinux上での解説を行います。
1、ダウンロードと解凍
こちらからLAMMPSの最新バージョンをダウンロードしてきます。
tarball(tarで圧縮されたアーカイブのこと)のファイル名にはバージョンが書かれていませんが、本テキストを書いている時点でのバージョンは22Jan14でした。
ダウンロードが完了したら、適当な場所に解凍します。
tar -xvzf lammps.tar.gz
cd lammps-22Jan14/
lammps-22Jan14をLAMMPSHOMEとし、特に記述がない限り、このディレクトリを基準にしているものとします。(例 cd src/はcd {LAMMPSを解凍したディレクトリ}/lammps-22Jan14/src/と言う意味)
2、パッケージの準備
*わりかし長いので、「パッケージなんて知るか」という方は読み飛ばして頂いても結構です。
LAMMPSでは本体のインストールの他に、Packageと呼ばれる補助的なプログラムを一緒にインストールすることができます。本体だけのインストールももちろん可能ですが、計算の高速化や様々な手法の適応など、便利なことが多いので、一緒にインストールしてみましょう。
まず、どんなパッケージが用意されているのかを確認します。
cd src/
make package-status // パッケージの一覧を表示することができます。
デフォルトではこんな感じでした。
Installed NO: package ASPHERE
Installed NO: package BODY
Installed NO: package CLASS2
Installed NO: package COLLOID
Installed NO: package DIPOLE
Installed NO: package FLD
Installed NO: package GPU
Installed NO: package GRANULAR
Installed NO: package KIM
Installed YES: package KSPACE
Installed YES: package MANYBODY
Installed NO: package MC
Installed NO: package MEAM
Installed NO: package MISC
Installed YES: package MOLECULE
Installed NO: package OPT
Installed NO: package PERI
Installed NO: package POEMS
Installed NO: package REAX
Installed NO: package REPLICA
Installed NO: package RIGID
Installed NO: package SHOCK
Installed NO: package SRD
Installed NO: package VORONOI
Installed NO: package XTC
Installed NO: package USER-ATC
Installed NO: package USER-AWPMD
Installed NO: package USER-CG-CMM
Installed NO: package USER-COLVARS
Installed NO: package USER-CUDA
Installed NO: package USER-EFF
Installed NO: package USER-LB
Installed NO: package USER-MISC
Installed NO: package USER-OMP
Installed NO: package USER-MOLFILE
Installed NO: package USER-PHONON
Installed NO: package USER-REAXC
Installed NO: package USER-SPH
USER-XXXと書いていないパッケージが” Standard Package ”と呼ばれるもので、基本的にはこの一連のパッケージさえインストールしてしまえば問題はないとおもいます。もちろん、Standard Packageを全てインストールする必要はないので、取捨選択しても構いません。USER-XXXと書いているパッケージは” User-contributed package “と呼ばれるもので、COLVARSやCUDAなど有用なパッケージも多いのですが、今回は簡単のためインストールはしていません。各パッケージの説明はこちらをご覧ください。なお、Installed YESと書いているパッケージはインストールを行う、Installed NOと書いているパッケージはインストールを行わないものを表します。
パッケージは何も考えずにインストールできるものもあれば、KIMやVORONOIのように外部プログラムを使用するために別途そのプログラムをインストールしておく必要があるもや、POEMSやMEAMのようにソースコードは提供されているが事前にビルドが必要なものなどもあります。まずはビルドから行ってみましょう。
2-1, lib中のビルド
*GPUは面倒臭くなったので説明やめました。パッケージも下の方で外しています。
cd lib/meam/
make -f Makefile.gfortran
cd lib/poems/
make -f Makefile.g++
cd lib/reax/
make -f Makefile.gfortran
長官たん超簡単ですね。いずれのMakeでもErrorが出ていないことを確認してください。何かトラブルがあった場合や、intel compilerでビルドを行いたい場合は、各サブディレクトリにあるREADMEファイルを読みましょう。Error messageでググると大抵はLAMMPSのMailing listで誰かが質問していて、Axelさんが「マニュアル読め」と怒っているのが見られると思います。
2.2.1 KIMの準備
こちらのページからいちばん新しいAPI(本テキストの時点では1.3.1)をダウンロードします。
例えばホームディレクトリで解凍したとすると、以下のようになります。
cd /home/qolo/
gzip -dc openkim-api-v1.3.1.tgz | tar xvf -
解凍したディレクトリであるopenkim-api-v1.3.1/に入り、
cd Makefile.KIM_Config.example Makefile.KIM_Config
Makefile.KIM_Configを例えば次のように修正しました。
KIM_DIR = /home/qolo/openkim-api-v1.3.1
Configは個別の環境に対応すること。例えばapple OSを使用している場合はこちらを。こちらの公式HPも参考に。
設定が終了したら、
export KIM_DIR=/home/qolo/openkim-api-v1.3.1
cd $KIM_DIR
make examples
make
なんか失敗したら、make cleanなどで一度キレイにしてから再度makeを行いましょう。
あとは{LAMMPSHOME}/src/KIM/Makefile.lammpsのkim_SYSINCとkim_SYSPATHを以下のように書き換えればOK!
kim_SYSINC = -I/home/qolo/openkim-api-v1.3.1/KIM_API
kim_SYSLIB = -lkim
kim_SYSPATH = -L/home/qolo/openkim-api-v1.3.1/KIM_API
2.2.2 VORONOIの準備
http://math.lbl.gov/voro++/download/このページからいちばん新しいvoro++(本テキストの時点では0.4.6)をダウンロードします。
今回もホームディレクトリに解凍します。
cd ~/
gzip -dc voro++-0.4.6.tar.gz | tar xvf -
管理者権限を持っていない場合は自分のホームディレクトリ以下にインストールすることになると思います。インストール先のディレクトリを作成しましょう(/usr/localなどにインストールする場合は以下の操作は必要ありません)
mkdir voro_dir //ディレクトリ名はなんでも構いません。
voro++のデフォルトのインストール先は/usr/local/だったような気がするので、voro++の設定ファイルを書き換え、インストール先を変更します。
cd ~/voro++-0.4.6/
vi config.mk
PREFIX=/home/qolo/voro_dir //インストール先のディレクトリを指定。
make
make install
次に、{LAMMPS HOME}/src/VORONOI/Makefile.lammpsを以下のように変更します。
(voro++のインストール先がデフォルトの場合は必要ありません。)
voronoi_SYSINC = -I/home/qolo/voro_dir/include/voro++
voronoi_SYSLIB = -lvoro++
voronoi_SYSPATH = -L/home/qolo/voro_dir/lib
これでパッケージの準備は完了です。ようやくLAMMPSのインストール・・・というところですが、その前にMPICHやFFTWの準備を行いましょう。
3、MPICHの準備
大抵の人はLAMMPSなどでMD計算を行う際、マルチコアで実行すると思います。もちろんシングルコアでも計算出来なくはないのですが、計算速度の面から実用的とは言い難いため、マルチコアでの計算が出来るような環境を整えておきましょう。
並列化計算のための規格として有名なのがMPIとOpenMPだと思います。Lammpsではどちらの並列化規格にも対応しており、もし好みがあるならば片方を選んでもいいと思います。が、私の経験上(そして周りの意見でも)MPIの方が断然速いので、本テキストではMPIを使用します。蛇足ですけど、OpenMPとOpen MPIは違うものなので、混同しないように。
本テキストではMPIによって並列計算を行う実装系としてMPICHを用います。MPI規格のOpenMPIも用いることが可能で、Axelさんはそっちのがおすすめみたいで、確かに速い気がします。Make.openmpiを用いれば結構アッサリインストール可能。詳述はしませんけど。
MPICHはLAMMPSに入っているわけではないので、そのインストールから始めることにしましょう。今回はmpich 3.0.4で行いました。MPICH2を使っても問題なく出来ました(前はうまくできなかったけど、最近やってみるとアッサリ)。MPICHのインストールの仕方はこちらなどを参照するといいと思います。ちなみに今回は/home/qolo/mpich_dir/以下をprefixしてインストールしました。
次に{LAMMPS HOME}/src/MAKE/Makefile.linux(iccがない場合はMakefile.g++)を以下のように編集します(バックアップとっておくのを忘れずに)。
MPI_INC = -I/home/qolo/mpich_dir/include
MPI_PATH = -L/home/qolo/mpich_dir/lib
MPI_LIB = -lmpich -lmpl -lfmpich -lpthread
4、fftw3の準備
インストールやFFTWの説明についてはhttp://www.kkaneko.com/rinkou/linux/fftw3linux.htm...のページが素晴らしいので基本的にそちらを踏襲。確かGromacsのインストールの時にも使用したと思いますし、結構いろいろな計算プログラムで用いるのでインストールされているかもしれません。ちなみに設定は./configure --prefix=/home/qolo/fftw3 --enable-float --enable-debugで行いました。
5、LAMMPSインストール
LAMMPSのインストールに必要なソフトのインストールが完了したら{LAMMPS HOME}/src/MAKE/Makefile.linuxを以下のように編集します。
FFT_INC = -I/home/qolo/fftw3/include
FFT_PATH = -L/home/qolo/fftw3/lib
FFT_LIB = -lfftw3f
ついでにJPG_LIBと-LAMMPS_JPEGも外しておきました。
これでようやく準備が整ったので、LAMMPSのインストールを始めます。まず2で折角準備したので、パッケージのインストール手続きから始めましょう。
cd src/
make yes-standard
これでstandard packageが全部インストールされる設定になりました。make package-statusと打って、確認してみましょう。
Installed YES: package ASPHERE
Installed YES: package BODY
Installed YES: package CLASS2
Installed YES: package COLLOID
Installed YES: package DIPOLE
Installed YES: package FLD
Installed YES: package GPU
src/pair_lj_sdk_coul_long_gpu.cpp does not exist
src/pair_lj_sdk_gpu.cpp does not exist
src/pair_lj_sf_dipole_sf_gpu.cpp does not exist
src/pair_lj_sdk_coul_long_gpu.h does not exist
src/pair_lj_sdk_gpu.h does not exist
src/pair_lj_sf_dipole_sf_gpu.h does not exist
Installed YES: package GRANULAR
Installed YES: package KIM
Installed YES: package KSPACE
Installed YES: package MANYBODY
Installed YES: package MC
Installed YES: package MEAM
Installed YES: package MISC
Installed YES: package MOLECULE
Installed YES: package OPT
Installed YES: package PERI
Installed YES: package POEMS
Installed YES: package REAX
Installed YES: package REPLICA
Installed YES: package RIGID
Installed YES: package SHOCK
Installed YES: package SRD
Installed YES: package VORONOI
Installed YES: package XTC
Installed NO: package USER-ATC
Installed NO: package USER-AWPMD
Installed NO: package USER-CG-CMM
Installed NO: package USER-COLVARS
Installed NO: package USER-CUDA
Installed NO: package USER-EFF
Installed NO: package USER-LB
Installed NO: package USER-MISC
Installed NO: package USER-OMP
Installed NO: package USER-MOLFILE
Installed NO: package USER-PHONON
Installed NO: package USER-REAXC
Installed NO: package USER-SPH
GPUパッケージについては2でも触れたとおりなにもしていないので文句をいわれてしまいます。このままLAMMPSのインストールを行っても途中なところでエラーを起こして止まってしまいますので、GPUを外しておきましょう。
make no-GPU
これで全ての準備は完了しました。いよいよインストールを行います。
make linux // MAKE/Makefile.XXのXXの部分に相当。Makefile.g++を使う場合、make g++とする。
LAMMPSのmakeはわりと時間がかかります。失敗して何か設定を変えて再挑戦する場合、機能を追加する場合などはmake clean-allで実行ファイルをクリーンな状態に戻してから再度makeを行いましょう。中途半端にコンパイルしたことによって整合性がとれなくなり、エラーなどの不具合が発生する可能性があります。
makeが成功すると、srcディレクトリの中にlmp_XX (XXはmake XXで指定したワードが入る。今回はmake linuxで行ったため、lmp_linuxが出来ている)が生成されます。これがLAMMPSの実行ファイルなので、パスが通してあるディレクトリに移すか、当該ディレクトリにパスを通しておきましょう。その前に、一応ちゃんとした実行ファイルが出来ているのかを確認するために、テストを行っておきます。
cp lmp_linux {LAMMPS HOME}/bench/
cd {LAMMPS HOME}/bench/
./lmp_linux < in.lj
ちゃんと実行ファイルが出来ていれば、計算結果が表示されるはずです。私の環境では以下の通りになりました。
LAMMPS (22 Jan 2014)
Lattice spacing in x,y,z = 1.6796 1.6796 1.6796
Created orthogonal box = (0 0 0) to (33.5919 33.5919 33.5919)
1 by 1 by 1 MPI processor grid
Created 32000 atoms
Setting up run ...
Memory usage per processor = 13.2266 Mbytes
Step Temp E_pair E_mol TotEng Press
0 1.44 -6.7733681 0 -4.6134356 -5.0197073
100 0.7574531 -5.7585055 0 -4.6223613 0.20726105
Loop time of 3.9805 on 1 procs for 100 steps with 32000 atoms
Pair time (%) = 3.42523 (86.0504)
Neigh time (%) = 0.410629 (10.316)
Comm time (%) = 0.0375006 (0.942109)
Outpt time (%) = 0.000181198 (0.00455215)
Other time (%) = 0.106955 (2.68696)
Nlocal: 32000 ave 32000 max 32000 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 19657 ave 19657 max 19657 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 1.20283e+06 ave 1.20283e+06 max 1.20283e+06 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 1202833
Ave neighs/atom = 37.5885
Neighbor list builds = 5
Dangerous builds = 0
ちなみにMPICHを用いて並列化すると、テスト計算のような小さな系でも威力を発揮します。8 coreで行ってみました。
~/mpich_dir/bin/mpirun -np 8 ./lmp_linux < in.lj // mpichもパスを通しておくことをお薦めします。
エネルギーなどはシングルコアと同じですので、計算時間のところだけ取り出すと、
Loop time of 0.680254 on 8 procs for 100 steps with 32000 atoms
Pair time (%) = 0.468042 (68.804)
Neigh time (%) = 0.0551136 (8.10192)
Comm time (%) = 0.143652 (21.1174)
Outpt time (%) = 7.51913e-05 (0.0110534)
Other time (%) = 0.013371 (1.96558)
となり、かなり速くなっていることが分かりました。MPIも無事動いているので、よかったよかった。8coreにしているのに1/5程度の短縮になっていますが、もっと長い計算を行うと、もっと並列化の威力を感じることが出来ると思います。
P.S. intel compilerも試してみましたが、MPICH系だと--enable-sharedオプションを入れてもShared Objectが見つからないとか言われてうまく行きませんでした。Openmpiならアッサリ行った気がするけど・・・http://sreedharmanchu.blogspot.jp/2011/08/lammps-f...を参考に、MPICCでコンパイルするとうまくいきました。ただし、当該ページはいらないオプションやたら多いし、記事自体も古いので注意。
''Cuda driver error 4 in call at file 'geryon/nvd_device.h' in line 124.''
This Error message distressed me ... :(
following text is very old version. you should see the newest official manual
本テキストはCentOS 5.10で行っています。gccなどの基本的なパッケージは最初から入っているため、以下では省いています。Windowsはバイナリが用意されていた(ような気がする)なので、OpenMPを使う場合はすぐに計算をすることができます(遅いけど)。Cygwin環境と併せていずれ機会があれば書いてみたいなぁとは思っていますが、今回はLinux上での解説を行います。
1、ダウンロードと解凍
こちらからLAMMPSの最新バージョンをダウンロードしてきます。
tarball(tarで圧縮されたアーカイブのこと)のファイル名にはバージョンが書かれていませんが、本テキストを書いている時点でのバージョンは22Jan14でした。
ダウンロードが完了したら、適当な場所に解凍します。
tar -xvzf lammps.tar.gz
cd lammps-22Jan14/
lammps-22Jan14をLAMMPSHOMEとし、特に記述がない限り、このディレクトリを基準にしているものとします。(例 cd src/はcd {LAMMPSを解凍したディレクトリ}/lammps-22Jan14/src/と言う意味)
2、パッケージの準備
*わりかし長いので、「パッケージなんて知るか」という方は読み飛ばして頂いても結構です。
LAMMPSでは本体のインストールの他に、Packageと呼ばれる補助的なプログラムを一緒にインストールすることができます。本体だけのインストールももちろん可能ですが、計算の高速化や様々な手法の適応など、便利なことが多いので、一緒にインストールしてみましょう。
まず、どんなパッケージが用意されているのかを確認します。
cd src/
make package-status // パッケージの一覧を表示することができます。
デフォルトではこんな感じでした。
Installed NO: package ASPHERE
Installed NO: package BODY
Installed NO: package CLASS2
Installed NO: package COLLOID
Installed NO: package DIPOLE
Installed NO: package FLD
Installed NO: package GPU
Installed NO: package GRANULAR
Installed NO: package KIM
Installed YES: package KSPACE
Installed YES: package MANYBODY
Installed NO: package MC
Installed NO: package MEAM
Installed NO: package MISC
Installed YES: package MOLECULE
Installed NO: package OPT
Installed NO: package PERI
Installed NO: package POEMS
Installed NO: package REAX
Installed NO: package REPLICA
Installed NO: package RIGID
Installed NO: package SHOCK
Installed NO: package SRD
Installed NO: package VORONOI
Installed NO: package XTC
Installed NO: package USER-ATC
Installed NO: package USER-AWPMD
Installed NO: package USER-CG-CMM
Installed NO: package USER-COLVARS
Installed NO: package USER-CUDA
Installed NO: package USER-EFF
Installed NO: package USER-LB
Installed NO: package USER-MISC
Installed NO: package USER-OMP
Installed NO: package USER-MOLFILE
Installed NO: package USER-PHONON
Installed NO: package USER-REAXC
Installed NO: package USER-SPH
USER-XXXと書いていないパッケージが” Standard Package ”と呼ばれるもので、基本的にはこの一連のパッケージさえインストールしてしまえば問題はないとおもいます。もちろん、Standard Packageを全てインストールする必要はないので、取捨選択しても構いません。USER-XXXと書いているパッケージは” User-contributed package “と呼ばれるもので、COLVARSやCUDAなど有用なパッケージも多いのですが、今回は簡単のためインストールはしていません。各パッケージの説明はこちらをご覧ください。なお、Installed YESと書いているパッケージはインストールを行う、Installed NOと書いているパッケージはインストールを行わないものを表します。
パッケージは何も考えずにインストールできるものもあれば、KIMやVORONOIのように外部プログラムを使用するために別途そのプログラムをインストールしておく必要があるもや、POEMSやMEAMのようにソースコードは提供されているが事前にビルドが必要なものなどもあります。まずはビルドから行ってみましょう。
2-1, lib中のビルド
*GPUは面倒臭くなったので説明やめました。パッケージも下の方で外しています。
cd lib/meam/
make -f Makefile.gfortran
cd lib/poems/
make -f Makefile.g++
cd lib/reax/
make -f Makefile.gfortran
長官たん超簡単ですね。いずれのMakeでもErrorが出ていないことを確認してください。何かトラブルがあった場合や、intel compilerでビルドを行いたい場合は、各サブディレクトリにあるREADMEファイルを読みましょう。Error messageでググると大抵はLAMMPSのMailing listで誰かが質問していて、Axelさんが「マニュアル読め」と怒っているのが見られると思います。
2.2.1 KIMの準備
こちらのページからいちばん新しいAPI(本テキストの時点では1.3.1)をダウンロードします。
例えばホームディレクトリで解凍したとすると、以下のようになります。
cd /home/qolo/
gzip -dc openkim-api-v1.3.1.tgz | tar xvf -
解凍したディレクトリであるopenkim-api-v1.3.1/に入り、
cd Makefile.KIM_Config.example Makefile.KIM_Config
Makefile.KIM_Configを例えば次のように修正しました。
KIM_DIR = /home/qolo/openkim-api-v1.3.1
Configは個別の環境に対応すること。例えばapple OSを使用している場合はこちらを。こちらの公式HPも参考に。
設定が終了したら、
export KIM_DIR=/home/qolo/openkim-api-v1.3.1
cd $KIM_DIR
make examples
make
なんか失敗したら、make cleanなどで一度キレイにしてから再度makeを行いましょう。
あとは{LAMMPSHOME}/src/KIM/Makefile.lammpsのkim_SYSINCとkim_SYSPATHを以下のように書き換えればOK!
kim_SYSINC = -I/home/qolo/openkim-api-v1.3.1/KIM_API
kim_SYSLIB = -lkim
kim_SYSPATH = -L/home/qolo/openkim-api-v1.3.1/KIM_API
2.2.2 VORONOIの準備
http://math.lbl.gov/voro++/download/このページからいちばん新しいvoro++(本テキストの時点では0.4.6)をダウンロードします。
今回もホームディレクトリに解凍します。
cd ~/
gzip -dc voro++-0.4.6.tar.gz | tar xvf -
管理者権限を持っていない場合は自分のホームディレクトリ以下にインストールすることになると思います。インストール先のディレクトリを作成しましょう(/usr/localなどにインストールする場合は以下の操作は必要ありません)
mkdir voro_dir //ディレクトリ名はなんでも構いません。
voro++のデフォルトのインストール先は/usr/local/だったような気がするので、voro++の設定ファイルを書き換え、インストール先を変更します。
cd ~/voro++-0.4.6/
vi config.mk
PREFIX=/home/qolo/voro_dir //インストール先のディレクトリを指定。
make
make install
次に、{LAMMPS HOME}/src/VORONOI/Makefile.lammpsを以下のように変更します。
(voro++のインストール先がデフォルトの場合は必要ありません。)
voronoi_SYSINC = -I/home/qolo/voro_dir/include/voro++
voronoi_SYSLIB = -lvoro++
voronoi_SYSPATH = -L/home/qolo/voro_dir/lib
これでパッケージの準備は完了です。ようやくLAMMPSのインストール・・・というところですが、その前にMPICHやFFTWの準備を行いましょう。
3、MPICHの準備
大抵の人はLAMMPSなどでMD計算を行う際、マルチコアで実行すると思います。もちろんシングルコアでも計算出来なくはないのですが、計算速度の面から実用的とは言い難いため、マルチコアでの計算が出来るような環境を整えておきましょう。
並列化計算のための規格として有名なのがMPIとOpenMPだと思います。Lammpsではどちらの並列化規格にも対応しており、もし好みがあるならば片方を選んでもいいと思います。が、私の経験上(そして周りの意見でも)MPIの方が断然速いので、本テキストではMPIを使用します。蛇足ですけど、OpenMPとOpen MPIは違うものなので、混同しないように。
本テキストではMPIによって並列計算を行う実装系としてMPICHを用います。MPI規格のOpenMPIも用いることが可能で、Axelさんはそっちのがおすすめみたいで、確かに速い気がします。Make.openmpiを用いれば結構アッサリインストール可能。詳述はしませんけど。
MPICHはLAMMPSに入っているわけではないので、そのインストールから始めることにしましょう。今回はmpich 3.0.4で行いました。MPICH2を使っても問題なく出来ました(前はうまくできなかったけど、最近やってみるとアッサリ)。MPICHのインストールの仕方はこちらなどを参照するといいと思います。ちなみに今回は/home/qolo/mpich_dir/以下をprefixしてインストールしました。
次に{LAMMPS HOME}/src/MAKE/Makefile.linux(iccがない場合はMakefile.g++)を以下のように編集します(バックアップとっておくのを忘れずに)。
MPI_INC = -I/home/qolo/mpich_dir/include
MPI_PATH = -L/home/qolo/mpich_dir/lib
MPI_LIB = -lmpich -lmpl -lfmpich -lpthread
4、fftw3の準備
インストールやFFTWの説明についてはhttp://www.kkaneko.com/rinkou/linux/fftw3linux.htm...のページが素晴らしいので基本的にそちらを踏襲。確かGromacsのインストールの時にも使用したと思いますし、結構いろいろな計算プログラムで用いるのでインストールされているかもしれません。ちなみに設定は./configure --prefix=/home/qolo/fftw3 --enable-float --enable-debugで行いました。
5、LAMMPSインストール
LAMMPSのインストールに必要なソフトのインストールが完了したら{LAMMPS HOME}/src/MAKE/Makefile.linuxを以下のように編集します。
FFT_INC = -I/home/qolo/fftw3/include
FFT_PATH = -L/home/qolo/fftw3/lib
FFT_LIB = -lfftw3f
ついでにJPG_LIBと-LAMMPS_JPEGも外しておきました。
これでようやく準備が整ったので、LAMMPSのインストールを始めます。まず2で折角準備したので、パッケージのインストール手続きから始めましょう。
cd src/
make yes-standard
これでstandard packageが全部インストールされる設定になりました。make package-statusと打って、確認してみましょう。
Installed YES: package ASPHERE
Installed YES: package BODY
Installed YES: package CLASS2
Installed YES: package COLLOID
Installed YES: package DIPOLE
Installed YES: package FLD
Installed YES: package GPU
src/pair_lj_sdk_coul_long_gpu.cpp does not exist
src/pair_lj_sdk_gpu.cpp does not exist
src/pair_lj_sf_dipole_sf_gpu.cpp does not exist
src/pair_lj_sdk_coul_long_gpu.h does not exist
src/pair_lj_sdk_gpu.h does not exist
src/pair_lj_sf_dipole_sf_gpu.h does not exist
Installed YES: package GRANULAR
Installed YES: package KIM
Installed YES: package KSPACE
Installed YES: package MANYBODY
Installed YES: package MC
Installed YES: package MEAM
Installed YES: package MISC
Installed YES: package MOLECULE
Installed YES: package OPT
Installed YES: package PERI
Installed YES: package POEMS
Installed YES: package REAX
Installed YES: package REPLICA
Installed YES: package RIGID
Installed YES: package SHOCK
Installed YES: package SRD
Installed YES: package VORONOI
Installed YES: package XTC
Installed NO: package USER-ATC
Installed NO: package USER-AWPMD
Installed NO: package USER-CG-CMM
Installed NO: package USER-COLVARS
Installed NO: package USER-CUDA
Installed NO: package USER-EFF
Installed NO: package USER-LB
Installed NO: package USER-MISC
Installed NO: package USER-OMP
Installed NO: package USER-MOLFILE
Installed NO: package USER-PHONON
Installed NO: package USER-REAXC
Installed NO: package USER-SPH
GPUパッケージについては2でも触れたとおりなにもしていないので文句をいわれてしまいます。このままLAMMPSのインストールを行っても途中なところでエラーを起こして止まってしまいますので、GPUを外しておきましょう。
make no-GPU
これで全ての準備は完了しました。いよいよインストールを行います。
make linux // MAKE/Makefile.XXのXXの部分に相当。Makefile.g++を使う場合、make g++とする。
LAMMPSのmakeはわりと時間がかかります。失敗して何か設定を変えて再挑戦する場合、機能を追加する場合などはmake clean-allで実行ファイルをクリーンな状態に戻してから再度makeを行いましょう。中途半端にコンパイルしたことによって整合性がとれなくなり、エラーなどの不具合が発生する可能性があります。
makeが成功すると、srcディレクトリの中にlmp_XX (XXはmake XXで指定したワードが入る。今回はmake linuxで行ったため、lmp_linuxが出来ている)が生成されます。これがLAMMPSの実行ファイルなので、パスが通してあるディレクトリに移すか、当該ディレクトリにパスを通しておきましょう。その前に、一応ちゃんとした実行ファイルが出来ているのかを確認するために、テストを行っておきます。
cp lmp_linux {LAMMPS HOME}/bench/
cd {LAMMPS HOME}/bench/
./lmp_linux < in.lj
ちゃんと実行ファイルが出来ていれば、計算結果が表示されるはずです。私の環境では以下の通りになりました。
LAMMPS (22 Jan 2014)
Lattice spacing in x,y,z = 1.6796 1.6796 1.6796
Created orthogonal box = (0 0 0) to (33.5919 33.5919 33.5919)
1 by 1 by 1 MPI processor grid
Created 32000 atoms
Setting up run ...
Memory usage per processor = 13.2266 Mbytes
Step Temp E_pair E_mol TotEng Press
0 1.44 -6.7733681 0 -4.6134356 -5.0197073
100 0.7574531 -5.7585055 0 -4.6223613 0.20726105
Loop time of 3.9805 on 1 procs for 100 steps with 32000 atoms
Pair time (%) = 3.42523 (86.0504)
Neigh time (%) = 0.410629 (10.316)
Comm time (%) = 0.0375006 (0.942109)
Outpt time (%) = 0.000181198 (0.00455215)
Other time (%) = 0.106955 (2.68696)
Nlocal: 32000 ave 32000 max 32000 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 19657 ave 19657 max 19657 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 1.20283e+06 ave 1.20283e+06 max 1.20283e+06 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 1202833
Ave neighs/atom = 37.5885
Neighbor list builds = 5
Dangerous builds = 0
ちなみにMPICHを用いて並列化すると、テスト計算のような小さな系でも威力を発揮します。8 coreで行ってみました。
~/mpich_dir/bin/mpirun -np 8 ./lmp_linux < in.lj // mpichもパスを通しておくことをお薦めします。
エネルギーなどはシングルコアと同じですので、計算時間のところだけ取り出すと、
Loop time of 0.680254 on 8 procs for 100 steps with 32000 atoms
Pair time (%) = 0.468042 (68.804)
Neigh time (%) = 0.0551136 (8.10192)
Comm time (%) = 0.143652 (21.1174)
Outpt time (%) = 7.51913e-05 (0.0110534)
Other time (%) = 0.013371 (1.96558)
となり、かなり速くなっていることが分かりました。MPIも無事動いているので、よかったよかった。8coreにしているのに1/5程度の短縮になっていますが、もっと長い計算を行うと、もっと並列化の威力を感じることが出来ると思います。
P.S. intel compilerも試してみましたが、MPICH系だと--enable-sharedオプションを入れてもShared Objectが見つからないとか言われてうまく行きませんでした。Openmpiならアッサリ行った気がするけど・・・http://sreedharmanchu.blogspot.jp/2011/08/lammps-f...を参考に、MPICCでコンパイルするとうまくいきました。ただし、当該ページはいらないオプションやたら多いし、記事自体も古いので注意。
コメントをかく