こんにちは、お久しぶりです。死んだと思ったかもしれませんが生きていました。qoloです。
最近なんかブログみたといってツイッターで連絡してくれる方もいて、嬉しい限りです。今後はちょいちょい更新していく可能性も否定できないということも吝かでないので、どうぞよろしくおねがいします。
以前、「インストールとテストの仕方は結構だが、ジョブをまともに流してないじゃないか!」という指摘を受けたので、今回はNAMDを使ったジョブの流し方、とある程度の解析について解説しようと思います。解析は次回になりますけど。それでは、はじまりはじまりー
vmdとNAMD をインストールする
http://mackerell.umaryland.edu/charmm_ff.shtmlからcharmmのForceField fileをダウンロードする。
それぞれ使えるようにする(省略)
PDBから2MT6のPDBファイルをダウンロードしてくる。まぁ、Human proteinの中で一番最近のやつだったからというだけで、何でもいいんですけどね。手元のマシンがクソしょぼいので、あんまりデカイのできないのでこの辺で。
NMRの構造なので、ゆらぎ分だけ複数の構造が含まれています。必要なのはひとつだけなので、VMDとかで取り出しておきましょうね。一応最終フレームの構造を取り出しておきました。
名前はそのままで、作業用フォルダ(/home/qolo/test/2MT6.pdb)に保存しておきました。
そいつを開いて、PSFファイルを作成します。これは分子量や電荷などの「原子の情報」と、結合様式が書かれているファイルです。PDBなどの座標データと、charmmのtopologyファイルから作成しましょう。今回はtop_all36_prot.rtfを使います。
まず、VMDでPDBファイルを読み込みます。それから、Extensions -> Modeling -> Automatic PSF Builderを選択
Topology filesのデフォルトの変なやつをDeleteし、top_all36_prot.rtfを選択。
Load input files -> Guess and split chains using current selections -> Create chains -> Apply patches and finish PSF/PDBで作成OKです。
次に、シミュレーションボックスに水を加えてやりましょう。
Extensions -> Modeling -> Add solvation box
・Waterbox Onlyのチェックボックスを外す。
・PSF,PDBに上で作ったファイル(デフォルトでは2MT6_autopsf.psfと2MT6_autopsf.pdb)を選択
・Boundary(間の距離の最小値)を2.0にします。デフォルトの2.4のままだと少々スッカスカなので。まぁ、計算機がいまいちという場合はそのままでもいいと思います。
・あとは今のところはデフォルトで、Use Molecule Dimensionsをチェックし、Box Paddingの欄に、全部5と代入。ざっくり言えば、タンパク質の端から5Åの大きさを持つボックスを作成する、という意味です。んで、Solvateをクリック。デフォルトの場合、solvate.pdbとsolvate.psfができます。
次に、系の電荷を中性化します。Extensions -> Add Ions
これはデフォルトのままで、AutoionizeでOK。勝手に中性化してくれます。今回の場合はClが一つ追加されて、溶媒分子がちょいちょいと変わったようです。電荷はほぼ0になりましたので、log画面で確認しておきましょう。
さて、いよいよ計算を流します。inputをチュートリアルとかから持ってきてもいいのですが、せっかくだから自分で作りましょう。最近のVMDにはインプット作成ツールまであります。便利なものですね。
今回は周期的境界条件(PBC)を使いたいので、セルの大きさなんかを定義してやります。なんとこれもツールでできます。ゴイス!
※使用するPBCToolsは、VMD1.9.1以下のバージョンで入っているものと、操作性がずいぶん違います、というか、重大なバグがあるらしいのでVMD1.9.2以上にはいっているPBCTools2.7以上を使用してください。
Extensions -> Tk Console
set cell [pbc get -now]
pbc writexst test.xst
これでセルの大きさが書かれたファイル(ionized.xst)が出来ました。
では、input fileを作っていきましょう!
Extensions -> Simulation -> NAMD Graphical Interface
Generalのところは適当に埋めて、Parameter filesをpar_all36_prot.prmとします。
あと、このパラメータセットにはTIP3P(水)のパラメータが入ってないので、param19.inpも入れておきましょう。
TimestepsのMolecular dynamicを、せっかくだから50,000にしてみましょう。後述するように2fs/stepにしているので、タイムスケールとしては100 psになります。
メニューバーからEdit -> Ensembleをクリック。なんとなくNPTにしました(通常はしばらくNPTで回してからNVT計算したりする。密度分かりませんからね。)。Particle Mesh Ewaldにもチェック。
Other Simulation parametersからtimestepを2、NonbondedFreqを1、FullElectFreqを2、StepPerCycleを10にします。あとはおこのみで。(画像参照)
大体設定できたら、Write NAMD config fileをクリック。適当なファイル名(デフォルトではionized.namd)になってると思います。
そうそう、今回はSHAKE法を使おうと思うので、fixedAtoms offの行を消してからionized.namdに以下の行を追加します。
rigidBonds all
rigidTolerance 5.0e-6
さて、では流そう・・・と思っても、大抵うまく行きません。特に中性化しちゃった夜なんかには。
par_all36_prot.prmの中身をちょっとイジりましょう。
ざっくりいうと、以下の二行を加えます。なお、これはtoppar_water_ions.strを参考にしました。
MASS 15 CLA 35.45000 CL ! Chloride Ion
CLA 0.0 -0.150 2.27 ! Chloride
(注釈:研究で実際に使うときは、ちゃんとForce Field Parameterは精査してください)
んで、コマンドにて(3コア使う場合)
charmrun namd2 +p3 ionized.namd > ionized.out
あとはしばらく待ちます。寝ながら。
というわけで、今日はここでおしまいです。また後日、しーゆー!
最近なんかブログみたといってツイッターで連絡してくれる方もいて、嬉しい限りです。今後はちょいちょい更新していく可能性も否定できないということも吝かでないので、どうぞよろしくおねがいします。
以前、「インストールとテストの仕方は結構だが、ジョブをまともに流してないじゃないか!」という指摘を受けたので、今回はNAMDを使ったジョブの流し方、とある程度の解析について解説しようと思います。解析は次回になりますけど。それでは、はじまりはじまりー
vmdとNAMD をインストールする
http://mackerell.umaryland.edu/charmm_ff.shtmlからcharmmのForceField fileをダウンロードする。
それぞれ使えるようにする(省略)
PDBから2MT6のPDBファイルをダウンロードしてくる。まぁ、Human proteinの中で一番最近のやつだったからというだけで、何でもいいんですけどね。手元のマシンがクソしょぼいので、あんまりデカイのできないのでこの辺で。
NMRの構造なので、ゆらぎ分だけ複数の構造が含まれています。必要なのはひとつだけなので、VMDとかで取り出しておきましょうね。一応最終フレームの構造を取り出しておきました。
名前はそのままで、作業用フォルダ(/home/qolo/test/2MT6.pdb)に保存しておきました。
そいつを開いて、PSFファイルを作成します。これは分子量や電荷などの「原子の情報」と、結合様式が書かれているファイルです。PDBなどの座標データと、charmmのtopologyファイルから作成しましょう。今回はtop_all36_prot.rtfを使います。
まず、VMDでPDBファイルを読み込みます。それから、Extensions -> Modeling -> Automatic PSF Builderを選択
Topology filesのデフォルトの変なやつをDeleteし、top_all36_prot.rtfを選択。
Load input files -> Guess and split chains using current selections -> Create chains -> Apply patches and finish PSF/PDBで作成OKです。
次に、シミュレーションボックスに水を加えてやりましょう。
Extensions -> Modeling -> Add solvation box
・Waterbox Onlyのチェックボックスを外す。
・PSF,PDBに上で作ったファイル(デフォルトでは2MT6_autopsf.psfと2MT6_autopsf.pdb)を選択
・Boundary(間の距離の最小値)を2.0にします。デフォルトの2.4のままだと少々スッカスカなので。まぁ、計算機がいまいちという場合はそのままでもいいと思います。
・あとは今のところはデフォルトで、Use Molecule Dimensionsをチェックし、Box Paddingの欄に、全部5と代入。ざっくり言えば、タンパク質の端から5Åの大きさを持つボックスを作成する、という意味です。んで、Solvateをクリック。デフォルトの場合、solvate.pdbとsolvate.psfができます。
次に、系の電荷を中性化します。Extensions -> Add Ions
これはデフォルトのままで、AutoionizeでOK。勝手に中性化してくれます。今回の場合はClが一つ追加されて、溶媒分子がちょいちょいと変わったようです。電荷はほぼ0になりましたので、log画面で確認しておきましょう。
さて、いよいよ計算を流します。inputをチュートリアルとかから持ってきてもいいのですが、せっかくだから自分で作りましょう。最近のVMDにはインプット作成ツールまであります。便利なものですね。
今回は周期的境界条件(PBC)を使いたいので、セルの大きさなんかを定義してやります。なんとこれもツールでできます。ゴイス!
※使用するPBCToolsは、VMD1.9.1以下のバージョンで入っているものと、操作性がずいぶん違います、というか、重大なバグがあるらしいのでVMD1.9.2以上にはいっているPBCTools2.7以上を使用してください。
Extensions -> Tk Console
set cell [pbc get -now]
pbc writexst test.xst
これでセルの大きさが書かれたファイル(ionized.xst)が出来ました。
では、input fileを作っていきましょう!
Extensions -> Simulation -> NAMD Graphical Interface
Generalのところは適当に埋めて、Parameter filesをpar_all36_prot.prmとします。
あと、このパラメータセットにはTIP3P(水)のパラメータが入ってないので、param19.inpも入れておきましょう。
TimestepsのMolecular dynamicを、せっかくだから50,000にしてみましょう。後述するように2fs/stepにしているので、タイムスケールとしては100 psになります。
メニューバーからEdit -> Ensembleをクリック。なんとなくNPTにしました(通常はしばらくNPTで回してからNVT計算したりする。密度分かりませんからね。)。Particle Mesh Ewaldにもチェック。
Other Simulation parametersからtimestepを2、NonbondedFreqを1、FullElectFreqを2、StepPerCycleを10にします。あとはおこのみで。(画像参照)
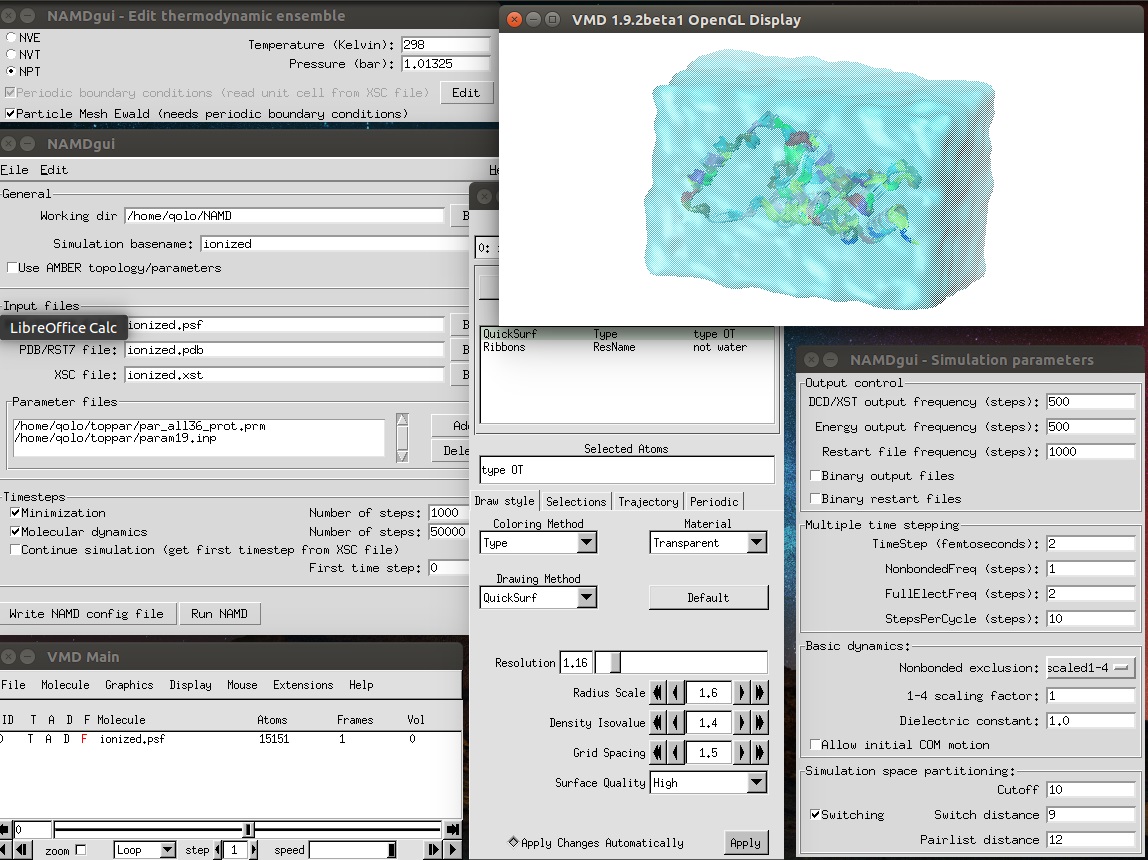
大体設定できたら、Write NAMD config fileをクリック。適当なファイル名(デフォルトではionized.namd)になってると思います。
そうそう、今回はSHAKE法を使おうと思うので、fixedAtoms offの行を消してからionized.namdに以下の行を追加します。
rigidBonds all
rigidTolerance 5.0e-6
さて、では流そう・・・と思っても、大抵うまく行きません。特に中性化しちゃった夜なんかには。
par_all36_prot.prmの中身をちょっとイジりましょう。
ざっくりいうと、以下の二行を加えます。なお、これはtoppar_water_ions.strを参考にしました。
MASS 15 CLA 35.45000 CL ! Chloride Ion
CLA 0.0 -0.150 2.27 ! Chloride
(注釈:研究で実際に使うときは、ちゃんとForce Field Parameterは精査してください)
んで、コマンドにて(3コア使う場合)
charmrun namd2 +p3 ionized.namd > ionized.out
あとはしばらく待ちます。寝ながら。
というわけで、今日はここでおしまいです。また後日、しーゆー!
コメントをかく